Staff profile
Professor Mark Wilson
Professor

| Affiliation | Telephone |
|---|---|
| Professor in the Department of Chemistry | +44 (0) 191 33 42144 |
Biography
Research Interests
We use classical molecular dynamics and Monte Carlo simulation methods to study problems in soft matter chemistry. We concentrate on materials that are ordered on the nanoscale, including liquid crystals, colloids, polymers, amphiphilic molecules and proteins. A key part of our work involves multiscale simulations of soft matter systems, including developing and testing new coarse-grained models.
Read about our latest simulations at my research group web pages.
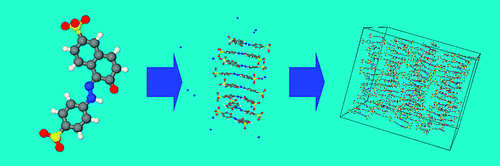
Coarse-grained simulations
We are developing new coarse-grained models for soft matter systems.1 Coarse-grained models allow molecular simulation to be carried out over time scales that are not accessible to conventional atomistic simulations. Long time scales allow us to observe self-assembly in solution, study polymers, observe segregation of molecules to an interface, or see formation of complex molecular structures mediated by specific molecular interactions.
Models to understand dynamic allostery in proteins
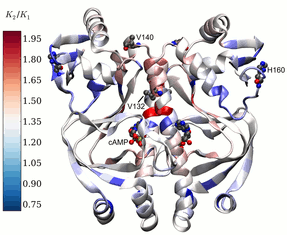
Allostery is a process by which a molecule binding to one site of a protein alters the activity of the protein at another site. We have developed new models to help understand how protein dynamics can influence allostery. In cases where the protein undergoes no conformational change on binding a small molecule, we demonstrate that that allostery arises as a natural consequence of changes in global low-frequency protein fluctuations on ligand binding.2
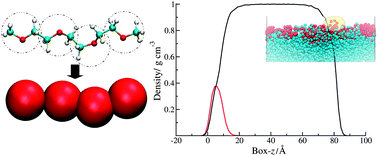
Simulation of chromonic liquid crystals
We have recently carried out the first detailed study of the structure and dynamics of a chromonic liquid crystal (edicol - the food dye sunset yellow).3 The simulations show spontaneous self-assembly in water at atomistic detail, and can be used to determine the free energy of association in solution. Sunset yellow is seen to form single molecule stacks with a preference for head-to-tail ordering, which at higher concentrations form liquid crystal phases in water (as shown in the picture above). Our work shows that simulation can be used to understand, predict and control molecular self-assembly with a view to the future use of chromonic materials for applications in self-assembled molecular electronics.
Dynamics in soft solids
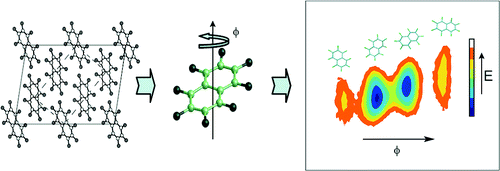
We have been using atomistic simulation to study dynamics in molecular solids.4 Our recent work with octafluoronaphthalene and urea inclusion compounds has indicated that its is now possible to look at dynamical processes over tens of ns, and study correlated motion in solids. This provides the possiblity of using simulation to complement and help interpret experimental solid state NMR data, and raises intriguing questions as to whether we can use simulation to help engineer tractable molecular machines for future applications.
Simulation of liquid crystal molecules and calculation of material properties
Molecular simulations provide a way of understanding the structure and dynamics5 of complex materials such as liquid crystals. We have recently used6 atomistic simulation to study the structure of the first low molecular weight biaxial nematic liquid crystal, ODBP-Ph-C7.
We have also been developing techniques to simulate liquid crystal molecules at the atomistic level and predict material properties in liquid crystal phases from simulation. For example, the rotational viscosity determines how quickly a nematic liquid crystal can reorientate in a liquid crystal display; and can be determined from our simulations by monitoring the liquid crystal director in the course of a molecular dynamics simulation.7 To carry out this work, we need accurate representations of intramolecular interactions within a molecule and intermolecular interactions between molecules. This is made possible by a new force field for use in materials chemistry, which we have been developing.8

The picture shows a sequence of time frames showing the slow growth of a biaxial phase from a uniaxial starting point. As the phase grows it develops ferroelectric domains, which are shown colour-coded. Molecules which are blue and red have transverse dipoles pointing in opposite directions.
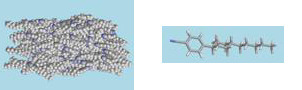
A snapshot from a molecular dynamics simulation showing the liquid crystal molecule PCH5 in a nematic phase.
Simulation studies of amphiphilic molecules in water
We are interested in the properties of amphiphilic molecules and have used simulation to understand the behaviour of amphiphilic polymers at a water/air interface. The simulations can be used to interpret the results of complementary neutron diffraction studies by calculating neutron reflectivity curves to match with experiment.9
Similar modelling techniques are being used to study lipid - amino acid interactions. Our recent atomistic work in this area seeks to understand how these interactions (specifically the balance of hydrogen bonding and cation-Pi interactions) can be perturbed by small chemical modifications of the amino acid structure. Such studies seek to aid our understanding of binding between proteins and cell membranes.
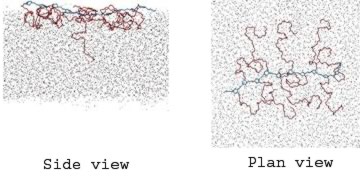
Snapshots from a simulation study of an amphiphilic polymer at a water-air interface at low surface concentration. The hydrophobic polynorbornene backbone is shown in blue and the hydrophilic poly(ethylene oxide) (PEO) grafts are shown in red. The dots represent water molecules. At higher surface concentrations the PEO chains are forced down into the aqueous subphase to form a polymer brush.
Coarse-grained models for the simulation of polymers, dendrimers, liquid crystals and lipids
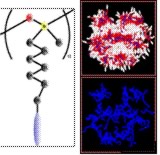
We have developed new coarse-grained models to study the bulk structure of liquid crystalline polymers and dendrimers10 and are working on “molecular engineering” tools to design molecules to have the desired organisation at the nanoscale. This research is being extended to lipid bilayers and vesicles. An important part of our work has involved developing efficient simulation methods that make use of parallel computers, so we are able to simulate extremely large systems composed of many molecules and take advantage of today’s supercomputers.11
The figure shows microphase separation in a liquid crystalline side chain polymer to give polymer-rich and liquid crystal-rich regions. Blue: polymer backbone (methylsiloxane), white: liquid crystal group, red: flexible spacer groups (alkyl chain).
More on Soft Matter Simulations
Read about our latest simulations at my research group web pages

References
- Prasitnok K., and Wilson M. R. A coarse-grained model for polyethylene glycol in bulk water and at a water/air interface. Phys. Chem. Chem. Phys., 2013, 15, 17093-1710.
- Rodgers T. L., Townsend P. D., Burnell D., Jones M. L., Richards S. A., McLeish T. C. B., Pohl E., Wilson M. R., Cann M. J. Modulation of Global Low-Frequency Motions Underlies Allosteric Regulation: Demonstration in CRP/FNR Family Transcription Factors. PLoS Biol, 2013, 11(9): e1001651.
- Chami F. and Wilson M. R. Molecular Order in a Chromonic Liquid Crystal: A Molecular Simulation Study of the Anionic Azo Dye Sunset Yellow. J. Am. Chem. Soc., 2010, 132, 7794.
- Ilott A. J., Palucha S., Batsanov A. S., Wilson M. R. and Hodgkinson P. Elucidation of Structure and Dynamics in Solid Octafluoronaphthalene from Combined NMR, Diffraction, And Molecular Dynamics Studies. J. Am. Chem. Soc., 2010, 132, 5179.
- Oganesyan, V. S., Kuprusevicius, E., Gopee, H., Cammidge, A. N., Wilson, M. R. Simulation of EPR spectra directly from molecular dynamics trajectories of a liquid crystal with doped paramagnetic spin probe. Phys. Rev. Lett., 2009, 102, 013005(1)-013005(4).
- Pelaez J. and Wilson M. R. Atomistic Simulations of a Thermotropic Biaxial Liquid Crystal. Phys. Rev. Lett., 2006, 97., 267801.
- Cheung D. L., Clark S. J, Wilson M. R. Calculation of the rotational viscosity of a nematic liquid crystal. Chem. Phys. Lett., 2002, 356, 140.
- Cheung D. L., Clark S. J., Wilson M. R. Parametrization and validation of a force field for liquid-crystal forming molecules. Phys. Rev. E, 2002, 65,051709.
- Anderson P. M. and Wilson M. R. Molecular Dynamics Simulations of an Amphiphilic Graft Copolymer at a Water/Air Interface., 2004, J. Chem. Phys, 121, 8503.
- Wilson M. R., Ilnytskyi J. M., Stimson L. M., Computer simulations of a liquid crystalline dendrimer in liquid crystalline solvents. J. Chem. Phys., 2003, 119, 3509.
- Ilnytskyi J, Wilson M. R.. A domain decomposition molecular dynamics program for the simulation of flexible molecules with an arbitrary topology of Lennard-Jones and/or Gay-Berne sites. Comput. Phys. Comm., 2001, 134, 23.
- Earl D. J., M., Wilson M. R. Predictions of molecular chirality and helical twisting powers: A theoretical study. J. Chem. Phys., 2003, 119, 10280.
Research interests
- Theoretical and Computational Chemistry
- Soft Matter
- Liquid Crystals and Polymers
Publications
Chapter in book
- Calculations of nematic mesophase properties using realistic potentials.Wilson, M. (2001). Calculations of nematic mesophase properties using realistic potentials. In D. Dunmur, A. Fukada, & G. Luckhurst (Eds.), Physical properties of liquid crystals: nematics. (pp. 635-643). Institution of Electrical engineers, England 2001.
Journal Article
- Polyesters incorporating biobased monomers for soil-release treatment of synthetic textile surfacesFiandra, E. F., Starck, M., Findlay, E. K., Binks, J., Si, G., Chilton, R., Sivik, M. R., Thompson, R. L., Wilson, M. R., & Mahon, C. S. (2025). Polyesters incorporating biobased monomers for soil-release treatment of synthetic textile surfaces. Green Chemistry, 27(44), 14290-14300. https://doi.org/10.1039/d5gc04056f
- Developing Multi-Component Solid Formulation Strategies for PROTAC Dissolution Enhancement.Screen, M. A., Askin, S., McCabe, J. F., Jacobs, E., Anane-Adjei, A., Mahon, C. S., Wilson, M. R., & Steed, J. W. (2025). Developing Multi-Component Solid Formulation Strategies for PROTAC Dissolution Enhancement. Molecular Pharmaceutics, 22(11), 6395-7150. https://doi.org/10.1021/acs.molpharmaceut.5c01107
- Crystallizing the Uncrystallizable: Insights from Extensive Screening of PROTACsScreen, M. A., McCabe, J. F., Askin, S., Guest, J. L., Hodgkinson, P., Cruz-Cabeza, A. J., Blundell, T. J., Rainer, D. N., Coles, S. J., Longcake, A., Probert, M. R., Mahon, C. S., Wilson, M. R., & Steed, J. W. (2025). Crystallizing the Uncrystallizable: Insights from Extensive Screening of PROTACs. Journal of the American Chemical Society, 147(31), 28056-28072. https://doi.org/10.1021/jacs.5c07977
- An unexpected chlorination of an organic sunscreenSmith, L. G., Di Leva, D., Shaw, L. A., Hughes, E., Kenwright, A. M., Beeby, A., Wilson, M. R., & Mahon, C. S. (2025). An unexpected chlorination of an organic sunscreen. Chemical Communications, 61(51), 9314-9317. https://doi.org/10.1039/d5cc01593f
- Designing lenalidomide cocrystals with an extended-release profile for improved pulmonary drug deliveryScreen, M. A., Tomkinson, G., McCabe, J. F., Askin, S., Mahon, C. S., Wilson, M. R., & Steed, J. W. (2025). Designing lenalidomide cocrystals with an extended-release profile for improved pulmonary drug delivery. New Journal of Chemistry, 49(16), 6535-6543. https://doi.org/10.1039/d5nj00425j
- Impact of charge distribution on the stability of ferroelectric nematic liquid crystalsde Mello, M., Wilson, M. R., & Araki, T. (2025). Impact of charge distribution on the stability of ferroelectric nematic liquid crystals. Soft Matter, 21(8), 1479-1488. https://doi.org/10.1039/d4sm01292e
- Surface Modification of Polyesters Using Biosourced Soil-Release PolymersStarck, M., Fiandra, E. F., Binks, J., Si, G., Chilton, R., Sivik, M., Thompson, R. L., Li, J., Wilson, M. R., & Mahon, C. S. (2025). Surface Modification of Polyesters Using Biosourced Soil-Release Polymers. JACS Au, 5(2), 666-674. https://doi.org/10.1021/jacsau.4c00908
- Dissipative particle dynamics parametrisation using infinite dilution activity coefficients: the impact of bondingHendrikse, R. L., Amador, C., & Wilson, M. R. (2025). Dissipative particle dynamics parametrisation using infinite dilution activity coefficients: the impact of bonding. Physical Chemistry Chemical Physics, 27(3), 1554-1566. https://doi.org/10.1039/d4cp03791j
- The essential synergy of MD simulation and NMR in understanding amorphous drug formsGuest, J. L., Bourne, E. A., Screen, M. A., Wilson, M. R., Pham, T. N., & Hodgkinson, P. (2025). The essential synergy of MD simulation and NMR in understanding amorphous drug forms. Faraday Discussions, 255, 325-341. https://doi.org/10.1039/d4fd00097h
- DPD simulations of anionic surfactant micelles: a critical role for polarisable water modelsHendrikse, R. L., Amador, C., & Wilson, M. R. (2024). DPD simulations of anionic surfactant micelles: a critical role for polarisable water models. Soft Matter, 20(37), 7521-7534. https://doi.org/10.1039/d4sm00873a
- Many-body dissipative particle dynamics simulations of micellization of sodium alkyl sulfatesHendrikse, R. L., Amador, C., & Wilson, M. R. (2024). Many-body dissipative particle dynamics simulations of micellization of sodium alkyl sulfates. Soft Matter, 20(30), 6044-6058. https://doi.org/10.1039/d4sm00533c
- Unravelling Guest Dynamics in Crystalline Molecular Organics Using 2 H Solid-State NMR and Molecular Dynamics SimulationErastova, V., Evans, I. R., Glossop, W. N., Guryel, S., Hodgkinson, P., Kerr, H. E., Oganesyan, V. S., Softley, L. K., Wickins, H. M., & Wilson, M. R. (2024). Unravelling Guest Dynamics in Crystalline Molecular Organics Using 2 H Solid-State NMR and Molecular Dynamics Simulation. Journal of the American Chemical Society, 146(27), 18360-18369. https://doi.org/10.1021/jacs.4c03246
- Computational predictions of interfacial tension, surface tension, and surfactant adsorption isothermsLi, J., Amador, C., & Wilson, M. R. (2024). Computational predictions of interfacial tension, surface tension, and surfactant adsorption isotherms. Physical Chemistry Chemical Physics, 26(15), 12107-12120. https://doi.org/10.1039/d3cp06170a
- An Electrochemistry and Computational Study at an Electrified Liquid–Liquid Interface for Studying Beta-Amyloid AggregationSilwane, B., Wilson, M., & Kataky, R. (2023). An Electrochemistry and Computational Study at an Electrified Liquid–Liquid Interface for Studying Beta-Amyloid Aggregation. Membranes, 13(6), Article 584. https://doi.org/10.3390/membranes13060584
- A many-body dissipative particle dynamics parametrisation scheme to study behaviour at air–water interfacesHendrikse, R., Amador, C., & Wilson, M. R. (2023). A many-body dissipative particle dynamics parametrisation scheme to study behaviour at air–water interfaces. Soft Matter, 19(20), 3590-3604. https://doi.org/10.1039/d3sm00276d
- Investigating anionic surfactant phase diagrams using dissipative particle dynamics: development of a transferable modelGray, S. J., Walker, M., Hendrikse, R., & Wilson, M. R. (2023). Investigating anionic surfactant phase diagrams using dissipative particle dynamics: development of a transferable model. Soft Matter, 19(17), 3092-3103. https://doi.org/10.1039/d2sm01641a
- Aided- and self-assembly of the liquid crystalline nanoparticles in bulk and in solution: computer simulation studiesSlyusarchuk, A., Yaremchuk, D., Lintuovori, J., Wilson, M., Grenzer, M., Sokolowski, S., & Ilnytskyi, J. (2023). Aided- and self-assembly of the liquid crystalline nanoparticles in bulk and in solution: computer simulation studies. Liquid Crystals, 50(1), 74-97. https://doi.org/10.1080/02678292.2023.2169872
- Computer Simulations of a Twist Bend Nematic (NTB): A Coarse-Grained Simulation of the Phase Behaviour of the Liquid Crystal Dimer CB7CBWilson, M. R., & Yu, G. (2023). Computer Simulations of a Twist Bend Nematic (NTB): A Coarse-Grained Simulation of the Phase Behaviour of the Liquid Crystal Dimer CB7CB. Crystals, 13(3). https://doi.org/10.3390/cryst13030502
- Molecular simulation approaches to the study of thermotropic and lyotropic liquid crystalsWilson, M. R., Yu, G., Potter, T. D., Walker, M., Gray, S. J., Li, J., & Boyd, N. J. (2022). Molecular simulation approaches to the study of thermotropic and lyotropic liquid crystals. Crystals, 12(5), Article 685. https://doi.org/10.3390/cryst12050685
- All-atom simulations of bent liquid crystal dimers: the twist-bend nematic phase and insights into conformational chiralityYu, G., & Wilson, M. R. (2022). All-atom simulations of bent liquid crystal dimers: the twist-bend nematic phase and insights into conformational chirality. Soft Matter, 18(15), 3087-3096. https://doi.org/10.1039/d2sm00291d
- Molecular simulation studies of self-assembly for a chromonic perylene dye: all-atom studies and new approaches to coarse-grainingGary, Y., & Wilson, M. R. (2022). Molecular simulation studies of self-assembly for a chromonic perylene dye: all-atom studies and new approaches to coarse-graining. Journal of Molecular Liquids, 345, Article 118210. https://doi.org/10.1016/j.molliq.2021.118210
- Phase behavior of correlated random copolymersPatyukova, E., Xi, E., & Wilson, M. (2021). Phase behavior of correlated random copolymers. Macromolecules, 54(6), 2763-2773. https://doi.org/10.1021/acs.macromol.0c02840
- Atomistic simulation studies of ionic cyanine dyes: self-assembly and aggregate formation in aqueous solutionYu, G., Walker, M., & Wilson, M. R. (2021). Atomistic simulation studies of ionic cyanine dyes: self-assembly and aggregate formation in aqueous solution. Physical Chemistry Chemical Physics, 23(11), 6408-6421. https://doi.org/10.1039/d0cp06205g
- Self-assembly and mesophase formation in a non-ionic chromonic liquid crystal: insights from bottom-up and top-down coarse-grained simulation modelsPotter, T. D., Walker, M., & Wilson, M. R. (2020). Self-assembly and mesophase formation in a non-ionic chromonic liquid crystal: insights from bottom-up and top-down coarse-grained simulation models. Soft Matter, 16(41), 9488-9498. https://doi.org/10.1039/d0sm01157f
- Oligomer/polymer blend phase diagram and surface concentration profiles for squalane/polybutadiene: experimental measurements and predictions from SAFT-g Mie and molecular dynamics simulationsTasche, J., Sabattie, E. F., Thompson, R. L., Campana, M., & Wilson, M. R. (2020). Oligomer/polymer blend phase diagram and surface concentration profiles for squalane/polybutadiene: experimental measurements and predictions from SAFT-g Mie and molecular dynamics simulations. Macromolecules, 53(7), 2309-2309. https://doi.org/10.1021/acs.macromol.9b02155
- Missing Link between Helical Nano‐ and Microfilaments in B4 Phase Bent‐Core Liquid Crystals, and Deciphering which Chiral Center Controls the Filament HandednessShadpour, S., Nemati, A., Salamończyk, M., Prévôt, M. E., Liu, J., Boyd, N. J., Wilson, M. R., Zhu, C., Hegmann, E., Jákli, A. I., & Hegmann, T. (2020). Missing Link between Helical Nano‐ and Microfilaments in B4 Phase Bent‐Core Liquid Crystals, and Deciphering which Chiral Center Controls the Filament Handedness. Small, 16(4), Article 1905591. https://doi.org/10.1002/smll.201905591
- Photoelectron spectroscopy of para-benzoquinone cluster anionsMensa-Bonsu, G., Wilson, M. R., Tozer, D. J., & Verlet, J. R. (2019). Photoelectron spectroscopy of para-benzoquinone cluster anions. Journal of Chemical Physics, 151(20), Article 204302. https://doi.org/10.1063/1.5132391
- Heliconical-layered nanocylinders (HLNCs) –hierarchical self-assembly in a unique B4 phase liquid crystal morphologyShadpour, S., Nemati, A., Boyd, N. J., Li, L., Prevot, M. E., Wakerlin, S. L., Vanegas, J. P., Salamonczyk, M., Hegmann, E., Zhu, C., Wilson, M. R., Jakli, A. I., & Hegmann, T. (2019). Heliconical-layered nanocylinders (HLNCs) –hierarchical self-assembly in a unique B4 phase liquid crystal morphology. Materials Horizons., 6(5), 959--968. https://doi.org/10.1039/c9mh00089e
- Assessing the transferability of common top-down and bottom-up coarse-grained molecular models for molecular mixturesPotter, T. D., Tasche, J., & Wilson, M. R. (2019). Assessing the transferability of common top-down and bottom-up coarse-grained molecular models for molecular mixtures. Physical Chemistry Chemical Physics, 21(4), 1912-1927. https://doi.org/10.1039/c8cp05889j
- Molecular simulation studies of cyanine-based chromonic mesogens: spontaneous symmetry breaking to form chiral aggregates and the formation of a novel lamellar structureThind, R., Walker, M., & Wilson, M. R. (2018). Molecular simulation studies of cyanine-based chromonic mesogens: spontaneous symmetry breaking to form chiral aggregates and the formation of a novel lamellar structure. Advanced Theory and Simulations., 1(10), Article 1800088. https://doi.org/10.1002/adts.201800088
- Direct Prediction of EPR Spectra from Lipid Bilayers: Understanding Structure and Dynamics in Biological MembranesCatte, A., White, G. F., Wilson, M. R., & Oganesyan, V. S. (2018). Direct Prediction of EPR Spectra from Lipid Bilayers: Understanding Structure and Dynamics in Biological Membranes. ChemPhysChem, 19(17), 2183-2193. https://doi.org/10.1002/cphc.201800386
- Emergent chirality in achiral liquid crystals: insights from molecular simulation models of the behaviour of bent-core mesogensLintuvuori, J. S., Yu, G., Walker, M., & Wilson, M. R. (2018). Emergent chirality in achiral liquid crystals: insights from molecular simulation models of the behaviour of bent-core mesogens. Liquid Crystals, 45(13-15), 1996-2009. https://doi.org/10.1080/02678292.2018.1492037
- Antimicrobial action of the cationic peptide, chrysophsin-3: a coarse-grained molecular dynamics studyCatte, A., Wilson, M. R., Walker, M., & Oganesyan, V. S. (2018). Antimicrobial action of the cationic peptide, chrysophsin-3: a coarse-grained molecular dynamics study. Soft Matter, 14(15), 2796-2807. https://doi.org/10.1039/c7sm02152f
- Validating an optimized GAFF force field for liquid crystals: TNI predictions for bent-core mesogens and the first atomistic predictions of a dark conglomerate phaseBoyd, N. J., & Wilson, M. R. (2018). Validating an optimized GAFF force field for liquid crystals: TNI predictions for bent-core mesogens and the first atomistic predictions of a dark conglomerate phase. Physical Chemistry Chemical Physics, 20(3), 1485-1496. https://doi.org/10.1039/c7cp07496d
- Effect of terminal chain length on the helical twisting power in achiral bent-core molecules doped in a cholesteric liquid crystalKim, B., Walker, M., Jo, S., Wilson, M. R., Takezoe, H., & Choi, S. (2018). Effect of terminal chain length on the helical twisting power in achiral bent-core molecules doped in a cholesteric liquid crystal. RSC Advances, 8(3), 1292-1295. https://doi.org/10.1039/c7ra11589j
- Development of new coarse-grained models for chromonic liquid crystals: insights from top-down approachesPotter, T. D., Tasche, J., Barrett, E. L., Walker, M., & Wilson, M. R. (2017). Development of new coarse-grained models for chromonic liquid crystals: insights from top-down approaches. Liquid Crystals, 44(12-13), 1979-1989. https://doi.org/10.1080/02678292.2017.1342005
- Predicting Oligomer/Polymer Compatibility and its Impact on Nanoscale Segregation in Thin FilmsSabattié, E., Tasche, J., Wilson, M. R., Skoda, M. W., Hughes, A. V., Lindner, T., & Thompson, R. (2017). Predicting Oligomer/Polymer Compatibility and its Impact on Nanoscale Segregation in Thin Films. Soft Matter, 13(19), 3580-3591. https://doi.org/10.1039/c7sm00048k
- Molecular dynamics simulations of the structure and the morphology of graphene/polymer nanocompositesGüryel, S., Walker, M., Geerlings, P., De Proft, F., & Wilson, M. (2017). Molecular dynamics simulations of the structure and the morphology of graphene/polymer nanocomposites. Physical Chemistry Chemical Physics, 19, 12959-12969. https://doi.org/10.1039/c7cp01552f
- Enhancement of the helical twisting power with increasing the terminal chain length of nonchiral bent-core molecules doped in a chiral nematic liquid crystalJo, S., Kim, B., Jeon, S., Bae, J., Walker, M., Wilson, M., Choi, S., & Takezoe, H. (2017). Enhancement of the helical twisting power with increasing the terminal chain length of nonchiral bent-core molecules doped in a chiral nematic liquid crystal. RSC Advances, 7(4), 1932-1935. https://doi.org/10.1039/c6ra27158h
- Simulation insights into the role of antiparallel molecular association in the formation of smectic A phasesWalker, M., & Wilson, M. R. (2016). Simulation insights into the role of antiparallel molecular association in the formation of smectic A phases. Soft Matter, 12(43), 8876-8883. https://doi.org/10.1039/c6sm01920j
- Formation of complex self-assembled aggregates in non-ionic chromonics: dimer and trimer columns, layer structures and spontaneous chiralityWalker, M., & Wilson, M. R. (2016). Formation of complex self-assembled aggregates in non-ionic chromonics: dimer and trimer columns, layer structures and spontaneous chirality. Soft Matter, 12(41), 8588-8594. https://doi.org/10.1039/c6sm01669c
- Easy creation of polymeric systems for molecular dynamics with Assemble!Degiacomi, M., Erastova, V., & Wilson, M. (2016). Easy creation of polymeric systems for molecular dynamics with Assemble! Computer Physics Communications, 202, 304-309. https://doi.org/10.1016/j.cpc.2015.12.026
- Optimization of the GAFF Force Field to Describe Liquid Crystal Molecules: The Path to a Dramatic Improvement in Transition Temperature PredictionsBoyd, N. J., & Wilson, M. R. (2015). Optimization of the GAFF Force Field to Describe Liquid Crystal Molecules: The Path to a Dramatic Improvement in Transition Temperature Predictions. Physical Chemistry Chemical Physics, 17(38), 24851-24865. https://doi.org/10.1039/c5cp03702f
- The role of protein-ligand contacts in allosteric regulation of the Escherichia coli Catabolite Activator ProteinTownsend, P., Rodgers, T., Glover, L., Korhonen, H., Richards, S., Colwell, L., Pohl, E., Wilson, M., Hodgson, D., McLeish, T., & Cann, M. (2015). The role of protein-ligand contacts in allosteric regulation of the Escherichia coli Catabolite Activator Protein. Journal of Biological Chemistry, 290(36), 22225-22235. https://doi.org/10.1074/jbc.m115.669267
- Global low-frequency motions in protein allostery: CAP as a model systemTownsend, P., Rogers, T., Pohl, E., Wilson, M., McLeish, T., & Cann, M. (2015). Global low-frequency motions in protein allostery: CAP as a model system. Biophysical Reviews, 7(2), 175-182. https://doi.org/10.1007/s12551-015-0163-9
- A simple model for chromonic aggregationWalker, M., & Wilson, M. (2015). A simple model for chromonic aggregation. Molecular Crystals and Liquid Crystals, 612(1), 117-125. https://doi.org/10.1080/15421406.2015.1030580
- Thermodynamics of the self-assembly of non-ionic chromonic molecules using atomistic simulations. The case of TP6EO2M in aqueous solutionAkinshina, A., Walker, M., Wilson, M., Tiddy, G., Masters, A., & Carbone, P. (2015). Thermodynamics of the self-assembly of non-ionic chromonic molecules using atomistic simulations. The case of TP6EO2M in aqueous solution. Soft Matter, 11(4), 680-691. https://doi.org/10.1039/c4sm02275k
- Self-assembly and mesophase formation in a non-ionic chromonic liquid crystal system: insights from dissipative particle dynamics simulationsWalker, M., Masters, A., & Wilson, M. (2014). Self-assembly and mesophase formation in a non-ionic chromonic liquid crystal system: insights from dissipative particle dynamics simulations. Physical Chemistry Chemical Physics, 16(42), 23074-23081. https://doi.org/10.1039/c4cp03092c
- A coarse-grained model for polyethylene glycol in bulk water and at a water/air interfacePrasitnok, K., & Wilson, M. R. (2013). A coarse-grained model for polyethylene glycol in bulk water and at a water/air interface. Physical Chemistry Chemical Physics, 15(40), 17093-17104. https://doi.org/10.1039/c3cp52958d
- Well-Tempered Metadynamics as a Tool for Characterizing Multi-Component, Crystalline Molecular MachinesIlott, A. J., Palucha, S., Hodgkinson, P., & Wilson, M. R. (2013). Well-Tempered Metadynamics as a Tool for Characterizing Multi-Component, Crystalline Molecular Machines. Journal of Physical Chemistry B (Soft Condensed Matter and Biophysical Chemistry), 117(40), 12286-12295. https://doi.org/10.1021/jp4045995
- Allostery without conformation change: modelling protein dynamics at multiple scalesMcLeish, T. C., Rodgers, T., & Wilson, M. R. (2013). Allostery without conformation change: modelling protein dynamics at multiple scales. Physical Biology, 10(5). https://doi.org/10.1088/1478-3975/10/5/056004
- DDPT: a comprehensive toolbox for the analysis of protein motionRodgers Thomas, L., Burnell, D., Townsend, P. D., Pohl, E., Cann, M. J., Wilson, M. R., & McLeish, T. C. (2013). DDPT: a comprehensive toolbox for the analysis of protein motion. BMC Bioinformatics, 14(1), Article 183. https://doi.org/10.1186/1471-2105-14-183
- Modulation of Global Low-Frequency Motions Underlies Allosteric Regulation: Demonstration in CRP/FNR Family Transcription FactorsRodgers, T., Townsend, P., Burnell, D., Jones, M., Richards, S., McLeish, T., Pohl, E., Wilson, M., & Cann, M. (2013). Modulation of Global Low-Frequency Motions Underlies Allosteric Regulation: Demonstration in CRP/FNR Family Transcription Factors. PLoS Biology, 11(9), Article e1001651. https://doi.org/10.1371/journal.pbio.1001651
- Structural Properties of Carboxylic Acid Dimers Confined within the Urea Tunnel Structure: An MD Simulation StudyIlott, A., Palucha, S., Batsanov, A., Harris, K., Hodgkinson, P., & Wilson, M. (2011). Structural Properties of Carboxylic Acid Dimers Confined within the Urea Tunnel Structure: An MD Simulation Study. Journal of Physical Chemistry B (Soft Condensed Matter and Biophysical Chemistry), 115(12). https://doi.org/10.1021/jp110137h
- Molecular Order in a Chromonic Liquid Crystal: A Molecular Simulation Study of the Anionic Azo Dye Sunset YellowChami, F., & Wilson, M. (2010). Molecular Order in a Chromonic Liquid Crystal: A Molecular Simulation Study of the Anionic Azo Dye Sunset Yellow. Journal of the American Chemical Society, 132(22), 7794-7802. https://doi.org/10.1021/ja102468g
- Theory and computer simulation for the cubatic phase of cut spheresDuncan, P., Dennison, M., Masters, A., & Wilson, M. (2009). Theory and computer simulation for the cubatic phase of cut spheres. Physical Review . E, Statistical, Nonlinear, and Soft Matter Physics, 79(3), Article 031702. https://doi.org/10.1103/physreve.79.031702
- Electron Paramagnetic Resonance Spectra Simulation Directly from Molecular Dynamics Trajectories of a Liquid Crystal with a Doped Paramagnetic Spin ProbeOganesyan, V., Kuprusevicius, E., Gopee, H., Cammidge, A., & Wilson, M. (2009). Electron Paramagnetic Resonance Spectra Simulation Directly from Molecular Dynamics Trajectories of a Liquid Crystal with a Doped Paramagnetic Spin Probe. Physical Review Letters, 102(1), Article 013005. https://doi.org/10.1103/physrevlett.102.013005
- A coarse-grained simulation study of mesophase formation in a series of rod–coil multiblock copolymersLintuvuori, J., & Wilson, M. (2009). A coarse-grained simulation study of mesophase formation in a series of rod–coil multiblock copolymers. Physical Chemistry Chemical Physics, 11(12), 2116-2125. https://doi.org/10.1039/b818616b
- Atomistic simulations of a thermotropic biaxial liquid crystalPelaez, J., & Wilson, M. (2006). Atomistic simulations of a thermotropic biaxial liquid crystal. Physical Review Letters, 97(26), Article 267801. https://doi.org/10.1103/physrevlett.97.267801
- Molecular dynamics simulations of side chain liquid crystal polymer molecules in isotropic and liquid-crystalline meltsStimson, L., & Wilson, M. (2005). Molecular dynamics simulations of side chain liquid crystal polymer molecules in isotropic and liquid-crystalline melts. Journal of Chemical Physics, 123(3). https://doi.org/10.1063/1.1948376
- Induced and spontaneous deracemization in bent-core liquid crystal phases and in other phases doped with bent-core moleculesEarl, D., Osipov, M., Takezoe, H., Takanishi, Y., & Wilson, M. (2005). Induced and spontaneous deracemization in bent-core liquid crystal phases and in other phases doped with bent-core molecules. Physical Review E, 71(2).
- Developing a force field for simulation of poly(ethylene oxide) based upon ab initio calculations of 1,2-dimethoxyethaneAnderson, P., & Wilson, M. (2005). Developing a force field for simulation of poly(ethylene oxide) based upon ab initio calculations of 1,2-dimethoxyethane. Molecular Physics, 103(1), 89-97.
- Progress in computer simulations of liquid crystalsWilson, M. (2005). Progress in computer simulations of liquid crystals. International Reviews in Physical Chemistry, 24(3-4), 421-455.
- Coarse-grained simulation studies of a liquid crystal dendrimer: towards computational predictions of nanoscale structure through microphase separationHughes, Z., Wilson, M., & Stimson, L. (2005). Coarse-grained simulation studies of a liquid crystal dendrimer: towards computational predictions of nanoscale structure through microphase separation. Soft Matter, 1(6), 436-443. https://doi.org/10.1039/b511082c
- Calculation of flexoelectric coefficients for a nematic liquid crystal by atomistic simulationCheung, D., Clark, S., & Wilson, M. (2004). Calculation of flexoelectric coefficients for a nematic liquid crystal by atomistic simulation. Journal of Chemical Physics, 121(18), 9131-9139. https://doi.org/10.1063/1.1802231
- Molecular dynamics simulations of amphiphilic graft copolymer molecules at a water/air interfaceAnderson, P., & Wilson, M. (2004). Molecular dynamics simulations of amphiphilic graft copolymer molecules at a water/air interface. Journal of Chemical Physics, 121(17), 8503-8510.
- Calculations of helical twisting powers from intermolecular torquesEarl, D., & Wilson, M. (2004). Calculations of helical twisting powers from intermolecular torques. Journal of Chemical Physics, 120(20), 9679-9683.
- Predictions of molecular chirality and helical twisting powers: A theoretical studyEarl, D., & Wilson, M. (2003). Predictions of molecular chirality and helical twisting powers: A theoretical study. Journal of Chemical Physics, 119(19), 10280-10288. https://doi.org/10.1063/1.1617980
- Helical twisting power and scaled chiral indicesNeal, M., Solymosi, M., Wilson, M., & Earl, D. (2003). Helical twisting power and scaled chiral indices. Journal of Chemical Physics, 119(6), 3567-3573.
- Computer simulations of a liquid crystalline dendrimer in liquid crystalline solventsWilson, M., Ilnytskyi, J., & Stimson, L. (2003). Computer simulations of a liquid crystalline dendrimer in liquid crystalline solvents. Journal of Chemical Physics, 119(6), 3509-3515. https://doi.org/10.1063/1.1588292
- Monte Carlo simulations of an amphiphilic polymer at a hydrophobic/hydrophilic interfaceMiller, A., Wilson, M., Cook, M., & Richards, R. (2003). Monte Carlo simulations of an amphiphilic polymer at a hydrophobic/hydrophilic interface. Molecular Physics, 101(8), 1131-1138.
- Rotational viscosities of Gay-Berne mesogensCuetos, A., Ilnytskyi, J., & Wilson, M. (2002). Rotational viscosities of Gay-Berne mesogens. Molecular Physics, 100(24), 3839-3845.
- A domain decomposition molecular dynamics program for the simulation of flexible molecules of spherically-symmetrical and nonspherical sites. II. Extension to NVT and NPT ensemblesIlnytskyi, J., & Wilson, M. (2002). A domain decomposition molecular dynamics program for the simulation of flexible molecules of spherically-symmetrical and nonspherical sites. II. Extension to NVT and NPT ensembles. Computer Physics Communications, 148(1), 43-58.
- Parametrization and validation of a force field for liquid-crystal forming moleculesCheung, D., Clark, S., & Wilson, M. (2002). Parametrization and validation of a force field for liquid-crystal forming molecules. Physical Review E: Statistical Physics, Plasmas, Fluids, and Related Interdisciplinary Topics, 65(5, Part 1), Article 051709. https://doi.org/10.1103/physreve.65.051709
- Calculation of the rotational viscosity of a nematic liquid crystal.Cheung, D., Clark, S., & Wilson, M. (2002). Calculation of the rotational viscosity of a nematic liquid crystal. Chemical Physics Letters, 356(1-2), 140-146. https://doi.org/10.1016/s0009-2614%2802%2900380-9
- Scaled chiral indices for ferroelectric liquid crystalsSolymosi, M., Low, R., Grayson, M., Neal, M., Wilson, M., & Earl, D. (2002). Scaled chiral indices for ferroelectric liquid crystals. Ferroelectrics, 277, 483-490.
- Computer simulations of soft repulsive spherocylindersEarl, D., Ilnytskyi, J., & Wilson, M. (2001). Computer simulations of soft repulsive spherocylinders. Molecular Physics, 99(20), 1719-1726.
- Molecular models in computer simulation of liquid crystalsIlnytskyi, J. M., & Wilson, M. R. (2001). Molecular models in computer simulation of liquid crystals. Journal of Molecular Liquids, 92(1-2), 21-28.
- A domain decomposition molecular dynamics program for the simulation of flexible molecules with an arbitrary topology of Lennard-Jones and/or Gay-Berne sitesIlnytskyi, J., & Wilson, M. (2001). A domain decomposition molecular dynamics program for the simulation of flexible molecules with an arbitrary topology of Lennard-Jones and/or Gay-Berne sites. Computer Physics Communications, 134(1), 23-32. https://doi.org/10.1016/s0010-4655%2800%2900187-9
- Calculating the helical twisting power of chiral dopantsWilson, M. R., & Earl, D. J. (2001). Calculating the helical twisting power of chiral dopants. Journal of Materials Chemistry, 11(11), 2672-2677.
- The first thousand-molecule simulation of a mesogen at the fully atomistic levelCook, M. J., & Wilson, M. R. (2001). The first thousand-molecule simulation of a mesogen at the fully atomistic level. Molecular Crystals and Liquid Crystals, 363, 181-+.
- A molecular dynamics simulation study of dipole correlation in the isotropic phase of the mesogens me5NF and GGP5CICook, M. J., & Wilson, M. R. (2001). A molecular dynamics simulation study of dipole correlation in the isotropic phase of the mesogens me5NF and GGP5CI. Molecular Crystals and Liquid Crystals, 357, 127-+.
- Development of an all-atom force field for the simulation of liquid crystal molecules in condensed phases (LCFF)Cook, M. J., & Wilson, M. R. (2001). Development of an all-atom force field for the simulation of liquid crystal molecules in condensed phases (LCFF). Molecular Crystals and Liquid Crystals, 357, 149-165.
- Calculation of helical twisting power for liquid crystal chiral dopantsWilson, M., & Cook, M. (2000). Calculation of helical twisting power for liquid crystal chiral dopants. Journal of Chemical Physics, 122, 1560-1564.
- Molecular dynamics simulation of liquid crystal phases using atomistic potentialsWilson, M., McBride, C., & Howard, J. (1998). Molecular dynamics simulation of liquid crystal phases using atomistic potentials. Molecular Physics, 93(6), 955-964.
- Molecular Dynamics Simulations of Flexible Liquid Crystal Molecules Using a Gay Berne/Lennard-Jones ModelWilson, M. (1997). Molecular Dynamics Simulations of Flexible Liquid Crystal Molecules Using a Gay Berne/Lennard-Jones Model. Journal of Chemical Physics, 107, 8654-8663.
- Molecular modelling of liquid crystal systems: An internal coordinate Monte Carlo approachWilson, M. (1996). Molecular modelling of liquid crystal systems: An internal coordinate Monte Carlo approach. Liquid Crystals, 21, 437-447.